







miND® Spike-In for small RNA-sequencing:
miND® spike-ins ensure your small RNA-seq data is publication-ready – with absolute normalization and validated quality control in a single reagent.
1. Quality control: confirm the dynamic range and quantitativeness of your small RNA sequencing experiments.
2. Absolute normalization: convert read counts to copies/µl total RNA and reduce bias originating from variation in RNA composition between samples.
miND® Spike-In design and usability:
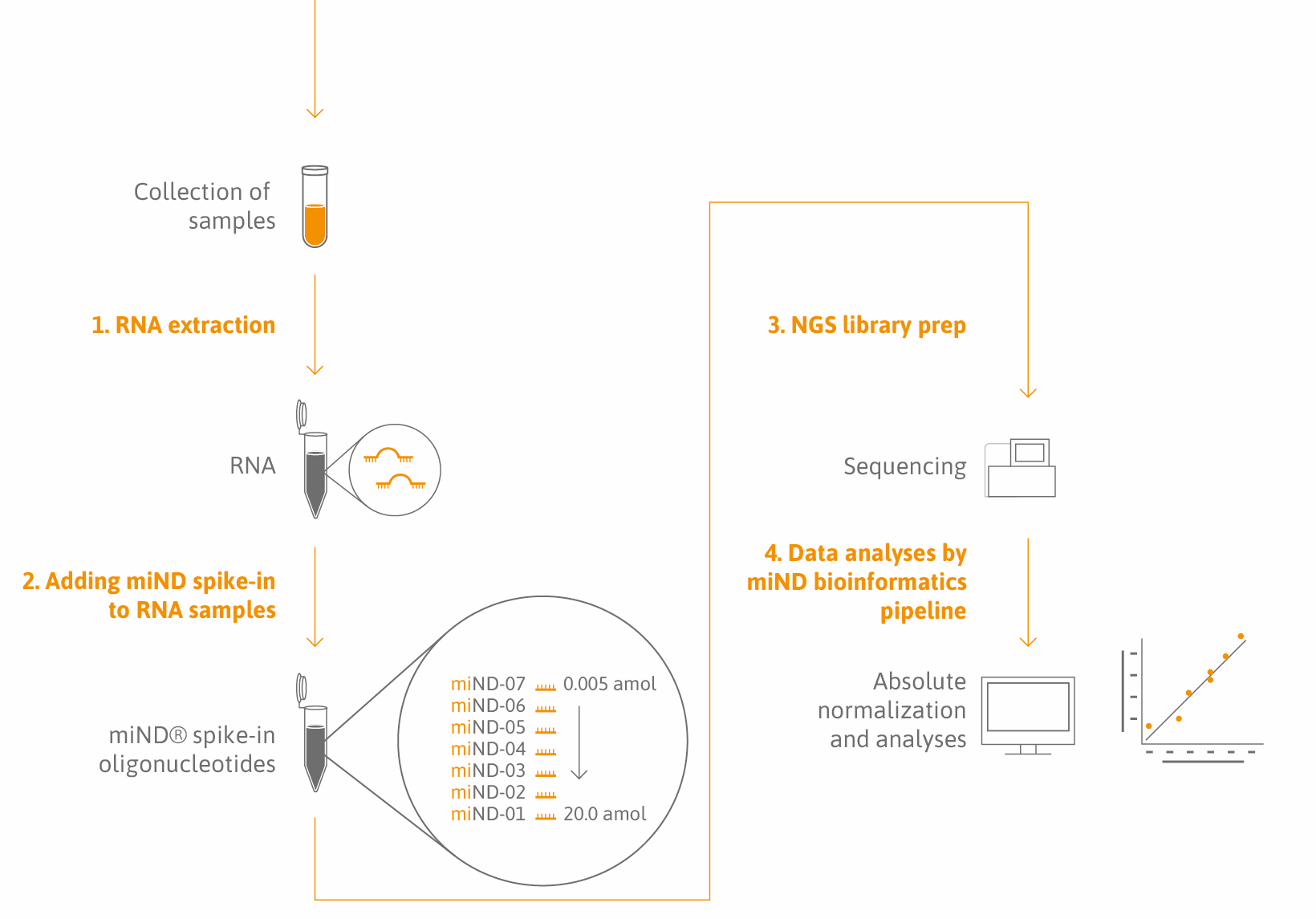
miND® Spike-Ins are a proprietary oligonucleotide mix with unique design features (Lutzmayer et al.). They are added to your total RNA samples before microRNA or small RNA sequencing analysis. The spike-in sequence composition and concentration range have been optimized to be compatible with almost any species and a broad range of sample types, including biofluids (serum, plasma, urine, CSF, synovial fluid), cells, tissues, extracellular vesicles, and non-vesicular fractions (Khamina et al.).


benefits
The challenge: small RNA-sequencing, especially for challenging input samples such as biofluids, exosomes, or low cell numbers, is prone to sequencing bias. In addition, the comparison of microRNAs and other small RNAs between sample types with differing RNA compositions is skewed based on the assumption of a constant amount of small RNAs per sample (the underlying hypothesis for reads per million (RPM) normalization).
Our solution: miND® Spike-Ins are a novel quality control parameter and normalizer that consists of seven oligonucleotides, each characterized by a unique core sequence flanked by 4 randomized nucleotides. miND® Spike-Ins are provided in a specific ratio to cover the broad concentration range of endogenous small RNAs.
Simplicity: miND® Spike-Ins come as ready-to-use reagents. No dilution, mixing, or change in your workflow is required – miND® spike-ins are directly added to your RNA sample and used for NGS library preparation.
Broad compatibility: miND® spike-ins have been successfully tested for their compatibility with a broad range of small RNA library preparation kits including RealSeq, QIASeq, miRVEL, NEXTFLEX TM v4, and NEBNext® Low-Bias.
Sample-to-insight: our public data analysis pipeline was specifically developed to convert small RNA-sequencing raw data into a simple but comprehensive report providing full access to your data in the conventional way (RPM, read count) as well as in absolute concentrations. On top of that, we have added several unsupervised and supervised analyses and QC parameters to the report.
Not familiar with small RNA-sequencing?
No problem – the entire miND small RNA-seq workflow is provided as a service by TAmiRNA – request a quotation.
price list
| Product Number | Formulation | Size | Shipment Conditions | Storage | Price* | Product Information | Order |
|---|---|---|---|---|---|---|---|
| KT-041-MIND-96 | lyophilized | 96 reactions | room temperature | lyophilized: store at -20°C or -80°C after reconstitution: store at -80°C | € 515 | product sheet / manual | request quote |
| KT-041-MIND-48 | resuspended ready-to-use | 48 reactions | dry ice | store at -80°C | € 279 | product sheet / manual | request quote |
* excluding shipment
product description
- 7 unique core sequences mixed in a specific proportions to cover a wide range of endogenous microRNAs
- Each unique core sequence consists of 13 nt that doesn’t match the genome of interest
- The core sequence is flanked by a set of 4 randomized nucleotides on both 5’ and 3’ ends
- Each of these 13 nt unique core sequences can be represented by up to 65,536 possible 21 nt sequences
- The miND spike-in contains up to 458,752 unique oligonucleotides
key publications and reference projects
Identification of miRNAs Present in Cell- and Plasma-Derived Extracellular Vesicles—Possible Biomarkers of Colorectal Cancer. Lenart M, Sieminska I, Szatanek R et al. Cancers (Basel). 2024 Jul 5;16(13):2464. doi: 10.3390/cancers16132464.
Analysis of extracellular vesicle microRNA profiles reveals distinct blood and lymphatic endothelial cell origins. Pultar M, Oesterreicher J, Hartmann J et al. J of Extracellular Bio.2024;3:e134; https://doi.org/10.1002/jex2.134
A MicroRNA Next-Generation-Sequencing Discovery Assay (miND) for Genome-Scale Analysis and Absolute Quantitation of Circulating MicroRNA Biomarkers Khamina K, Diendorfer A, Skalicky S, et al. Int. J. Mol. Sci. 2022, 23(3), 1226; https://doi.org/10.3390/ijms23031226
miND (miRNA NGS Discovery pipeline): a small RNA-seq analysis pipeline and report generator for microRNA biomarker discovery studies Diendorfer A, Khamina K, Pultar M and Hackl M. F1000Research 2022, 11:233, 1226; https://doi.org/10.12688/f1000research.94159.1
MicroRNA-30d-5p—A Potential New Therapeutic Target for Prevention of Ischemic Cardiomyopathy after Myocardial Infarction
Boxhammer E, Paar V, Wernly B et al. Cells 2023, 12(19), 2369; https://doi.org/10.3390/cells12192369
Unique miRNome and transcriptome profiles underlie microvascular heterogeneity in mouse kidney
Luxen M, Zwiers PJ, Meester F et al. Am J Physiol Renal Physiol. 2023 Sep 1;325(3):F299-F316. doi: 10.1152/ajprenal.00005.2023. Epub 2023 Jul 6.
Investigation of MicroRNA Biomarkers in Equine Distal Interphalangeal Joint Osteoarthritis
Baker ME., Lee S., Clinton M. et al. Int J Mol Sci 2022 Dec 8;23(24):15526. doi: 10.3390/ijms232415526.
Modulation of Host-Parasite Interactions with Small Molecules Targeting Schistosoma mansoni microRNAs
Hamway Y., Zimmermann K., Blommers MJJ. et al. ACS Infect Dis. 2022 Oct 14;8(10):2028-2034. doi: 10.1021/acsinfecdis.2c00360. Epub 2022 Sep 13.
Comprehensive Characterization of Platelet-Enriched MicroRNAs as Biomarkers of Platelet Activation
Krammer TL., Zeibig S., Schrottmaier WC. et al. Cells 2022 Apr 7;11(8):1254. doi: 10.3390/cells11081254.
The role of extracellular vesicle miRNAs and tRNAs in synovial fibroblast senescence
Wijesinghe SN., Anderson J., Brown TJ. et al. Front Mol Biosci. 2022 Sep 23;9:971621. doi: 10.3389/fmolb.2022.971621. eCollection 2022.
Small non-coding RNA landscape of extracellular vesicles from a post-traumatic model of equine osteoarthritis
Anderson JR., Jacobsen S., Walters M. et al. Front Vet Sci. 2022 Aug 8;9:901269. doi: 10.3389/fvets.2022.901269. eCollection 2022.
MicroRNA Expression Profiling in Porcine Liver, Jejunum and Serum upon Dietary DON Exposure Reveals Candidate Toxicity Biomarkers
Segura-Wang M. Grenier B., Ilic S. et al. Int J Mol Sci. 2021 Nov 7;22(21):12043. doi: 10.3390/ijms222112043./em>
Novel small RNA spike-in oligonucleotides enable absolute normalization of small RNA-Seq data Lutzmayer S, Enugutti B and Nodine MD. Sci Rep. 2017 Jul 19;7(1):5913. doi: 10.1038/s41598-017-06174-3.
Small non-coding RNA landscape of extracellular vesicles from a post-traumatic model of equine osteoarthritis Anderson JR., Jacobsen S., Walters M. et al. Front. Vet. Sci., 08 August 2022, Sec. Veterinary Regenerative Medicine, https://doi.org/10.3389/fvets.2022.901269
frequently asked questions (FAQ)
Product overview
Compatibility & Application
Yes. We have tested the compatibility and performance of the miND® spike-ins with several library preparation kits, including:
- RealSeq®-Biofluids Small RNA Library Preparation Kit (RealSeq Biosciences)
- QIAseq miRNA Library Kit (Qiagen)
- NEXTFLEX Small RNA Sequencing Kit v4 (Revvity)
- miRVEL Discovery Small RNA-Seq Library Prep (Lexogen)
- NEBNext® Small RNA Library Prep (New England Biolabs)
- NEBNext® Low-bias Small RNA Library Prep Kit (New England Biolabs)
This list reflects kits that feature different adapter ligation principles and that have been formally evaluated in combination with the miND® spike-in controls. Other small RNA library preparation protocols should also be compatible; however, performance validation data may not be available.
The spike-in concentration range has been selected to match the typical endogenous miRNA abundance in biofluid samples and have been extensively tested on:
- Biofluids: serum, plasma, cerebrospinal fluid, synovial fluid, urine, and saliva
- Extracellular vesicles and non-vesicular fractions extracted from biofluids and cell culture supernatant
For recommendations on how to use the spike-ins with cellular and tissue input, see sections below.
The spike-ins have been developed and optimized for use with biofluids and other low RNA input samples. The standard use is therefore for total RNA inputs ranging from 0.5 – 50 ng at which the addition of 1 µl spike-in is recommended. For use with inputs >50 ng total RNA the recommended spike-in volume scales with the total RNA input to ensure that the spike-in signals remain proportionate to the endogenous miRNA signal.
| Total RNA input | Recommended spike-in volume | Typical sample types |
|---|---|---|
| Standard: 0.5 – 50 ng | 1 µl | Biofluids (serum, plasma, urine, synovial fluid), extracellular vesicles |
| 50 – 100 ng | 1 – 2 µl | Cells, small tissue amounts |
| ≥ 100 ng | ≥ 2 µl | Tissue, high-input cell experiments |
Important: If you change the spike-in volume from the standard recommended 1 µl, you must update the corresponding volume in the absolute concentration calculations. See the next question for details.
Yes, this is critical if you intend to work with the spike-in normalized data. The absolute concentration calculation is based on the known input amount of each spike-in. When the volume added differs from the standard 1 µl, the actual amount of spike-in introduced into the reaction changes, and the calibration curve will be shifted accordingly.
Update the spike-in input volume parameter in your analysis to match the volume used. Failing to do so, will cause a proportional error in all calculated concentrations: e.g. using 2 µl without updating the calculation will cause concentrations to appear 2x lower than the true values.
If you are using the miND pipeline (TAmiRNA), the spike-in volume is a configurable input parameter in the markdown file.
Generally, no. The spike-ins are optimized for biofluid samples, which are inherently low-input matrices. The standard volume specified in the product´s IFU is designed for this range and should not require dilution for typical biofluid inputs and EVs.
The spike-ins are optimized for addition directly to the extracted RNA sample immediately before small RNA library preparation.
The spike-ins are optimized for addition directly to the extracted RNA sample prior to library preparation. While pre-isolation addition is possible, it requires additional consideration and is not the default recommendation.
As a rule of thumb, use the following approach to determine the appropriate spike-in volume:
- Identify the spike-in volume (Vspike-in (lib prep)) and RNA input volume (VRNA input) recommended in your library preparation protocol and sample type.
- Calculate the spike-in ratio: Divide the recommended spike-in volume by the RNA input volume.
- Multiply this ratio by your RNA elution volume (Velution).
- Apply a correction factor based on the estimated RNA extraction efficiency (E) of your workflow to obtain the pre-isolation spike-in volume (Vspike-in (pre-isolation)).

Example: The protocol recommends 1 µl spike-in added to 8.5 µl RNA input as starting material for library preparation. If RNA is eluted in 50 µl and extraction efficiency is estimated at 70%, the recommended pre-isolation volume would be (1/8.5) x 50 x (1/0.7) = 8.4 µl.
Important considerations:
- Add the spike-ins to the lysis buffer or lysis master mix prior to sample addition. Do not add the spike-ins directly into the biological sample (e.g. plasma).
Note: Spike-ins added directly to a biological sample are exposed to the sample matrix before protective lysis conditions are established. This leads to degradation by RNases present in the sample.
- If extraction efficiency is unknown, an estimate of 50-70% (E = 0.5-0.7) is a reasonable starting point.
- Optimization and pilot testing are strongly recommended before applying pre-isolation spike-in addition to a full experiment, as recovery can vary between extraction kits, sample matrices and library preparation methods.
- Absolute quantification is not recommended when spike-ins are added prior to RNA isolation as the fraction of spike-ins recovered is not precisely known.
Quality
Data Analysis
The following questions cover the bioinformatics workflow for processing miND® spike-in data. They complement the wet-lab guidance above and address the most frequent questions we receive about the open-source scripts and Docker tools published at https://github.com/tamirna.
Troubleshooting

